
1. Introduction
Virgin olive oil (VOO) is obtained exclusively from the fruit of the olive tree (Oleaeuropaea L.) using mechanical and physical means, such as washing, decantation, centrifugation, and filtration, means that do not lead to alterations in the oil, excluding of oils obtained using solvents or re-esterification processes and of any mixture with other oils [1].
According to the chemical composition, VOO can be divided into the saponifiable fraction, which represent more than 98-99% of total oil weight, such as free fatty acid (FA) and triacylglycerols (TAGs) and unsaponifiable fraction, about 1-2% of the oil weight (e.g., hydrocarbons, fatty alcohols) [2]. Minor compounds, such as, phenolics, vitamins, and volatile compounds among others, are the responsible of VOOs sensory characteristics, nutritional value and biological properties that can differentiate it from other vegetable fats [2]. Moreover of agronomical factors (such as geographical origins, variety, climatic conditions, agronomical practices, fruit ripeness, etc.), there are many technological factors that can affect the content of minor components, like the effects of the washing operation, crushing and malaxation conditions, separation system, clarification, storage or filtration system [3, 4].
The clarification step is responsible for removing the moisture and solid particle suspended in the oil from the horizontal screw solid bowl. The clarification devices used in VOO extraction are both vertical centrifugation and natural settling in tanks. If impurities remain, are not well removed before the oil storage, are responsible of anaerobic fermentation processes (hydrolysis and oxidation reactions). Fermentations affect the oil quality and chemical composition, including, sensory characteristics causing the emergence of undesired flavors typical of these processes, such as ‘fusty’ or ‘muddy’ sediment [5].
The main factors that influence these clarification systems are: the density difference between liquid phase and solid particles, the particle size, the liquid viscosity, etc. [6]. Therefore, knowing oil density and viscosity is essential for the design, sizing and selection of proper equipment for the olive oil industry, such as settling and centrifugation devices, including pumps, pipes, filtration systems, etc. In addition, both physicochemical properties, especially viscosity, are very sensitive to temperature [7]. In fact, while oil density decreases linearly with increasing temperature [7], viscosity follows the temperature dependence of the Arrhenius model [8-10]. On other hand, from the sensorial point of view, the viscosity can be associated with the term ‘fluidity’, a kinaesthetic characteristic of the rheological properties of the oil, the set of which are capable of stimulating the mechanical receptors located in the mouth during the test [11], where oil with low viscosity means a higher fluidity. Many oil tasters detect viscosity differences during VOO sensory evaluation from different olive cultivars. Although this topic is not included in the official method, these differences can be linked with the oil fatty acid composition [12].
Several authors have already studied the density and viscosity on vegetable oils, including olive oil [7, 8, 13]. Rodenbush et al. [13] developed a generalized method to estimate the liquid density of vegetable oils from fatty acid by a correlation based on fatty acid critical properties and composition oil. In this same work these authors also proposed a method to predict viscosity from density data, relating both physical properties. Many studies have been performed in order to find a correlation between viscosity and FA and/or TAG composition for different vegetable oils, although not only olive oil [8-10, 14]. In fact, physical properties, such as crystal structure, solubility, viscosity, and melting point, have been demonstrated to be affected by the position distribution of FA in TAG [15]. However, because olive oil shows a high variability in composition depending on agronomical and technological factors, the available data are still scarce.
Differential scanning calorimetry (DSC) is a thermoanalytical technique widely used for the evaluation of quality parameters of vegetable oil and fats due to its higher precision as well as the small amount of sample required [16-18]. Its application presents several advantages as it does not require chemical treatments or time- consuming manipulation practices before each measurement. Different vegetable oils showed complex thermal profiles that are mainly due to the great variety of TAG as their principal constituents [19]. In addition, a relationship between thermal properties measured by DSC have been found to be statistically correlated with chemical parameters related to composition for vegetable oils in the past [20-22], and more recently also evaluated in VOO characterization [23], taking into account transition upon cooling. Chiavaro et al. [24] have also used DSC measurements to determinate relation between thermal properties and chemical composition for the discrimination of commercial categories of olive oil. In addition, relationships between thermal properties and composition (FA and TAG) have been described for VOO samples from Spain [16], Tunisia [25] and Italy [26, 27]. In a recent work, Maggio et al. [28] have developed and validated a faster analytical method based on a HPLC-DSC and partial least square (PLS) procedure on a large set of VOO samples from different Mediterranean area in order to add new information about the influence of TAG composition on cooling profiles with the aim to enforce the application of DSC in the field of quality evaluation. On the other hand, Santos et al. [29] related thermoanalytical and kinetic properties of edible oils with theirs chemical compositions and viscosities as a function of temperature whereas Vecchio et al. [30] studied the thermoxidation of VOO by thermogravimety and DSC analysis relating thermal properties to chemical composition of several monovarietals VOOs.
In general, chemical methods are commonly applied for the evaluation of VOOs quality and thermal stability, but they are known to be expensive, to be time-consuming and to have a high environmental impact. Moreover, calorimetric techniques have demonstrated their validity as supporting tool in order to improve VOOs classification. The aim of this work is to study not only thermal properties from thermal profiles (crystallization, melting, and oxidation curves) by DSC measurements, but also physical characterization (density and viscosity) as a function of temperature of VOOs from 14 different olive cultivars, in order to relate thermal and physical properties with chemical composition (FA).
2. Materials and methods
2.1 Materials
In this study developed during 2012/13 crop year, 16 VOO samples from 14 monovarietal VOOs were used ‘Picual I’ (Pi-I), ‘Hojiblanca’ (Hj), ‘Arbequina’ (Ar), ‘Blanqueta I’ (Bl-I), ‘Blanqueta II’ (Bl-II), ‘Manzanilla Prieta’ (M.P), ‘Frantoio’ (Fr), ‘Moraiolo’ (Mo), ‘Verdial de Badajoz’ (V.B), ‘Tempranillo de Lucena’ (T.L), ‘Negrillo de la Carlota’ (N.C), ‘Konservolia’ (Kn), ‘Picual II’ (Pi-II), ‘Aloreña de Iznalloz’ (A.I), ‘Menara’ (Me), ‘Kalokerida’ (Kl), which were obtained from the Olive Germplasm Bank Collection of Cordoba (Spain). These oils were selected with the aim to cover the whole range of possible olive oil compositions taking into account the FA. Oil extraction was performed in IFAPA ‘Venta del Llano’ research center, Mengíbar (Jaén, Spain), using an Abencor laboratory oil mill (Abengoa, Seville), kneading the olive paste at 28°C for 30 min. Finally, the oil was filtered and stored at -24°C prior to perform any analysis.
2.2 Oil characterization
2.2.1 Fatty acid determination
Fatty acid methyl esters (FAMEs) composition was determined according to the European Regulation 2568/91 [31]. Chromatographic separation was performed on a Perkin-Elmer Clarus 400 (USA) gas chromatograph (GC) equipped with a split/splittless injector (temperature 250°C) and a FID detector (temperature 300°C). A BPX silica capillary column (SGE, Australia) (50 m x 0.22 mm id, 0.25 pm film thickness) was used. The oven temperature was held at 198°C and helium was used as carrier gas (Inlet pressure, 28 psi). The results were expressed as peak area (relative) percent.
2.2.2 Differential Scanning Calorimetry (DSC)
Oil samples (5-7 mg) were weighed in aluminum pans, covers were sealed into place and analyzed with a DSC822. (Mettler Toledo, Switzerland). Initially, oil samples were equilibrated at 25°C for 5 min, cooled to -80°C at a rate of 5 °C/min equilibrated at -80°C for 5 min, heated to 25°C at a rate of 5°C/min and finally, once equilibrated at 25°C for 1 min, heated from 25°C to 300 °C at a rate of 5°C/min. An air flow was used as gas purged at a flow rate of 100 mL/min. DSC curves were analyzed with STAR
Software (Versión 8.10, METTLER TOLEDO) to obtain enthalpy (AH, J/g), temperature of the major peak of crystallization phase (Pc, °C), temperature of the major peak of melting phase (Pm1,°C), temperature of the minor peak of melting phase (Pm2,°C), initial temperature of transition (to,°C), end temperature of transition (te, °C), range of the transition (R, °C, difference between to and te) and initial temperature of the oxidation curve (tox, °C). A single analysis was performed per oil.
2.2.3 Density determination
Olive oil density was determined (by duplicate) by Densito 30PX portable density meter (Mettler Toledo, Switzerland). Samples were introduced into a glass tube and equilibrated at different temperatures (10, 20, 30 and 40°C) during 15 min by using a Phoenix II P1-C25P refrigerated bath circulator (ThermoHaake, Germany) prior to perform density measurements. Results were expressed as g/cm3. The relationship between density and temperature was expressed mathematically by means of Eq. (1):
![]()
where p is the density expressed in g/cm3, T is the temperature expressed in °C, b is the intercept and m is the slope.
2.2.4 Rheological measurements
Dynamic viscosity was measured (by triplicate) by a controlled-strain rheometer (ARES) (TA Instruments, USA). The geometry used has been a cone and plate geometry (diameter: 50mm). Rheological tests were performed between 0.1 and 100 s-1 and carried out between 10 and 40°C at 5°C intervals (also measurements at 0 and 5°C were performed for the ‘Pi-I’, ‘Hj’, and ‘Ar’ samples). In addition, in order to ensure precise and stable control of temperature during measurements (±0.1°C) a programmable refrigerating and heating circulator (Julabo FS10 HD, Germany) was used. Results were reported as Pa ■ s. Arrhenius model (Eq. (2)) has been selected to describe the effect of temperature on VOO viscosity.
![]()
where p is the oil dynamic viscosity (Pa- s), A the pre-exponential factor (Pa – s), Ea is the activation energy (J/mol), R is the gas constant (8.314J/mol/K) and Tthe absolute temperature (K).
2.3 Statistical analysis
Means and standard deviations were calculated with statistical package Statistix, Version 9.1 (USA). The Statistix software was used to perform one-way analysis of variance (ANOVA) and Tukey’s honest significant difference test at a 95% confidence level (p< 0.005) to identify differences among groups. Pearson correlation coefficients (r) were calculated between DSC thermogram features and chemical parameters of oil samples, at a 95% confidence level (p<0.05). Oil samples were also discriminated by multivariate parametric methods where the principal component analysis (PCA) was carried out using The Unscrambler software package (Version 9.7, CAMO, Oslo, Norway).
3. Results and discussion
3.1 FAMEs composition of VOOs
Table 1 reports the content of the main FA for the sixteen VOOs (‘Pi-I’, ‘Hj’, ‘Ar’, ‘Bl- I’, ‘Bl-II’, ‘M.P’, ‘Mo’, ‘Fr’, ‘V.B’, ‘T.L’, ‘N.C’, ‘Kn’, ‘Pi-II’, ‘A.I’, ‘Me’, and ‘Kl’) which were also grouped in saturated FA (SFA), unsaturated FA (UFA), monounsaturated FA (MUFA), polyunsaturated FA (PUFA) and the MUFA/PUFA ratio. There was a high degree of variability in FA composition for the oils from different olive varieties although their values are similar to those published by other authors [16, 17]. The predominant FA are oleic acid (C18:1), which reaches values between 44.3 and 81% for the ‘Bl-I’ and ‘Kl’ varieties, respectively, followed by the palmitic (C16:0) and linoleic acids (C18:2). An exception was found for ‘A.I’, ‘Bl-I’, and ‘V.B’ varieties where the linoleic acid contents are higher than palmitic acid content. MUFA contents were ranged between 48.0 and 82.5% corresponding to ‘Bl-I’ and ‘Kl’ oil varieties, respectively. On the other hand, SFA content ranged between 12.1 and 25.7%, corresponding to ‘Bl-I’ and ‘Me’, respectively. SFA low content for ‘Bl-I’ oil is a logic result because of the high oleic and linoleic contents.
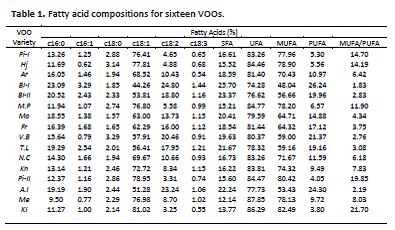
Fatty acids: c16:0 palmitic acid, c16:1 palmitoleic acid, c18:0 stearic acid, c18:1 oleic acid, c18:2 linoleic, c18:3 linolenic, SFA saturated fatty acid, UFA unsaturated fatty acid, MUFA monosaturated fatty acid, PUFA polyunsaturated fatty acid.
3.2 DSC analysis
Figure 1 exhibits DSC thermograms for the ‘Pi-I’, ‘Hj’, and ‘Ar’ samples: cooling curves (Fig. 1A), heating curves (Fig. 1B) and the initial curve of the oxidation process (Fig. 1C). In addition, thermal parameters were obtained from the thermograms for all VOOs (Table 2).
3.2.1. DSC analysis of cooling thermogram
Cooling curves from 25 to -80°C (Fig. 1A), showed a typical DSC cooling thermograms for this type of vegetable oil, as previously reported in literature [16, 17, 19, 23-25, 28], with two exothermic events, a well-defined main peak (Pc) and other secondary peak, not so well defined.
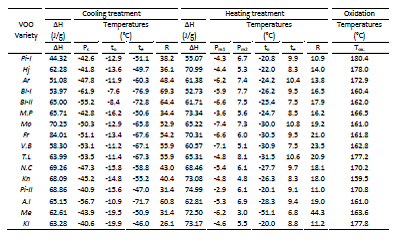
The crystallization peaks in the DSC thermograms showed an initial temperature of crystallization (to) with valúes from -19.9 to -7.6 °C, the peak for temperature of crystallization (Pc) achieved valúes between -61.9 and -40.6 °C, the peak for temperature of end crystallization (te) showed valúes ranged between -46.0 and -76.9°C, the peak area related to enthalpy of crystallization (AH) with values between 44.32 to 84.91J/g and the temperature difference between t0 and te (R) ranged from 26.1°C to 69.3°C. Differences for peak maximum and shapes were previously attributed to the difference in fatty acid composition/or initial oxidative status for this oil [16, 17, 26, 32], as shown in Fig. 1A where three quite different cooling curves were reported.
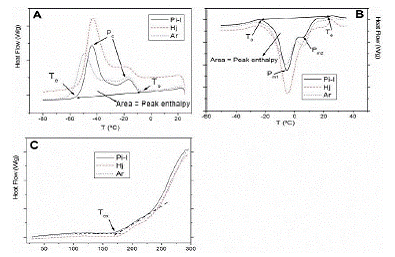
Figure 1. Representative DSC thermograms of three selected varieties: ‘Pi-I’, ‘Hj’, and ‘Ar’, (A) DSC thermogram during cooling from room temperature to -80°C, (B) DSC melting thermogram during heating from -80 to 25°C and (C) DSC oxidation thermogram during heating from 25 to 300°C. (Scanning rate of 5°C/min for the three thermograms).
3.2.2 DSC analysis of heating thermograms
Heating curves from -80 to 25°C (Fig. 1B), showed the heating profiles with an endothermic phase transition with two well defined peaks due to melting. Similar melting shapes were described in previous works [16, 17, 19, 25, 32, 33]. Table 2 showed the melting thermal parameters. The melting peaks in the DSC thermograms exhibited an initial melting temperature (t0) with valúes between -51.1 and -20.0°C for the ‘Me’ and ‘Kl’ varieties, respectively. The temperature of melting for the minor peak (Pm1) show values from -2.9 to -7.4 °C, corresponding to the ‘Pi-II’ and ‘Ma’ varieties, respectively. In general, can be observed that for varieties with higher PUFA content showed lower Pm1 values, similar behaviors were described by Jiménez et al. [16].The temperature of melting for the major peak (Pm2) was comprised between ‘Me’ (3.0°C) and ‘T.L’ (8.1°C) varieties. For the peak temperature of end melting (te), it showed values ranged between 6.8 and 10.8°C for the ‘Me’ and ‘Ma’ varieties, respectively. The peak area as enthalpy of melting (AH) values varied between 52.73 to 74.99 J/g corresponding to the ‘Pi-II’ and ‘Ma’ varieties, respectively. The temperature difference between to and te (R) ranged from 10.9°C (‘Pi-I’ variety) to 44.3°C (‘Me’ variety).
Table 2. DSC parameters obtained from transition phase of the thermograms for the sixteen VOOs. AH, enthalpy; Pc, temperature of the major peak of crystallization phase; Pm1, temperature of the major peak of melting phase; Pm2, temperature of the minor peak of melting phase; to and Te, initial and end temperature of the transition phase, respectively; R, range of the transition phase (temperature difference between t0 and te); Tox, initial temperature of the oxidation process.
3.2.3 DSC analysis of oxidation thermograms
For the analysis of the DSC oxidation process, that other authors have analyzed in more detail [30], only was studied the beginning of the process from 25 to 300°C. Figure 1C showed the initial curve of the oxidation process for the ‘Pi-I’, ‘Hj’, and ‘Ar’ samples. Two zones can be observed, the first with a slight increase of the curve slope, and other where occurs a sharp change of the slope with a great increment of the heat flow. Similar shapes for the beginning of the oxidation process were described previously [16, 30].Different initial temperatures of the oxidation process (íox) can be observed for each oil sample (Table 2), showing values comprised between 159.5 and 180.4°C for the ‘Kn’ and ‘Pi-I’ varieties, respectively.